Epigenetic Analysis of Mouse Brain Aging
An Integrated Epigenetic Analysis of Mouse Brain Aging Reveals Insights into Neurodegeneration: An integrated epigenetic analysis provides new insights into neurodegeneration by defining a comprehensive single-cell multi-omic atlas of the aging mouse brain.
Our Current Understanding of the Epigenetics of Aging in the Brain
The seemingly unstoppable progression of normal aging can lead to deleterious alterations in the human brain, and, as such, aging constitutes the primary risk factor for the development of multiple distinct neurodegenerative diseases (Hou et al.). Importantly, significant alterations at the epigenetic level also accompany aging (Lopez-Otin et al., Yang et al., and Lu et al.) and have been linked to neurodegeneration. But how? The loss of DNA methylation can induce the re-expression of transposable elements, which comprise a substantial fraction of mammalian genomes, promoting cellular dysfunction and age-related decline through increased genome instability and inflammation (Simon et al., De Cecco et al., and Della Valle et al.). In addition, disrupted genome stability and altered chromatin conformation also play important roles in neurodegeneration and aging (Dileep et al.).
Researchers led by Margarita Behrens and Joseph Ecker (Salk) aimed to move past recent studies that characterized cell-type-specific transcriptomic (Jin et al.) and select epigenetic alterations during brain aging (Chien et al., Zhang et al., Emani et al., and Tan et al.) and undertake a comprehensive multi-omic analysis across multiple brain regions to provide mechanistic insight into the region- and cell-type-specific effects of mouse aging. In their recent Cell study, Zeng et al. report the generation of a single-cell data set encompassing DNA methylation, chromatin conformation, and spatial transcriptomics, and the integration of this data with chromatin accessibility and transcriptome data (Amaral et al.) across brain regions vulnerable to neurodegeneration. Can these findings deepen our understanding of the processes involved in the aging brain and potentially identify targetable mechanisms that may help to treat age-associated neurodegenerative diseases?
Paired-Tag technology from Epigenome Technologies generates joint epigenetic and transcriptomic profiles at single-cell resolution and detects histone modifications and RNA transcripts in nuclei with efficiency comparable to single-nucleus RNA-seq/ChIP-seq assays. Could integrating Paired-Tag into the experimental scheme employed have provided even deeper single-cell epigenetic insight into the links between brain aging and the development of neurodegenerative diseases by analyzing histone modification profiles?
Integrative Epigenetic Analyses Underscore Age-related and Cell-specific Alterations to DNA Methylation, Chromatin Confirmation, and Gene Expression
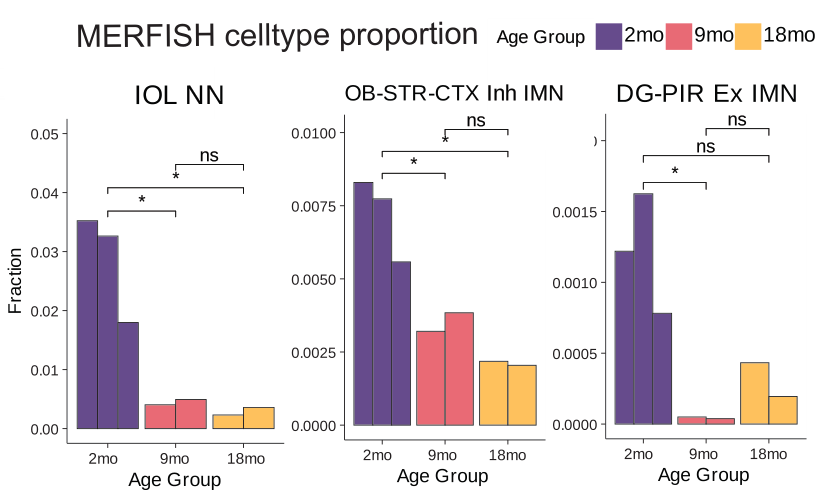
The single-nucleus cell atlas of brain aging across multiple brain regions developed in this study included over 130,000 single-cell methylomes and nearly 73,000 joint chromatin conformation-methylome nuclei and described thirty-six major cell types following integration with companion transcriptomic and chromatin accessibility data. The study profiled eight major brain regions across three ages (2, 9, and 18 months) in adult mice.
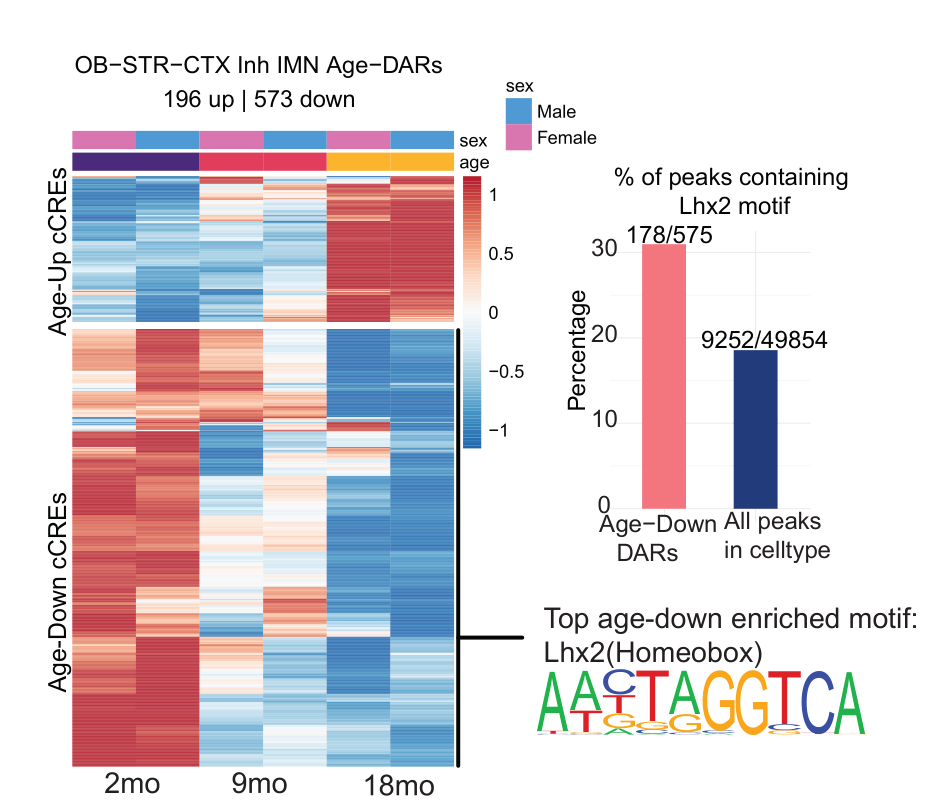
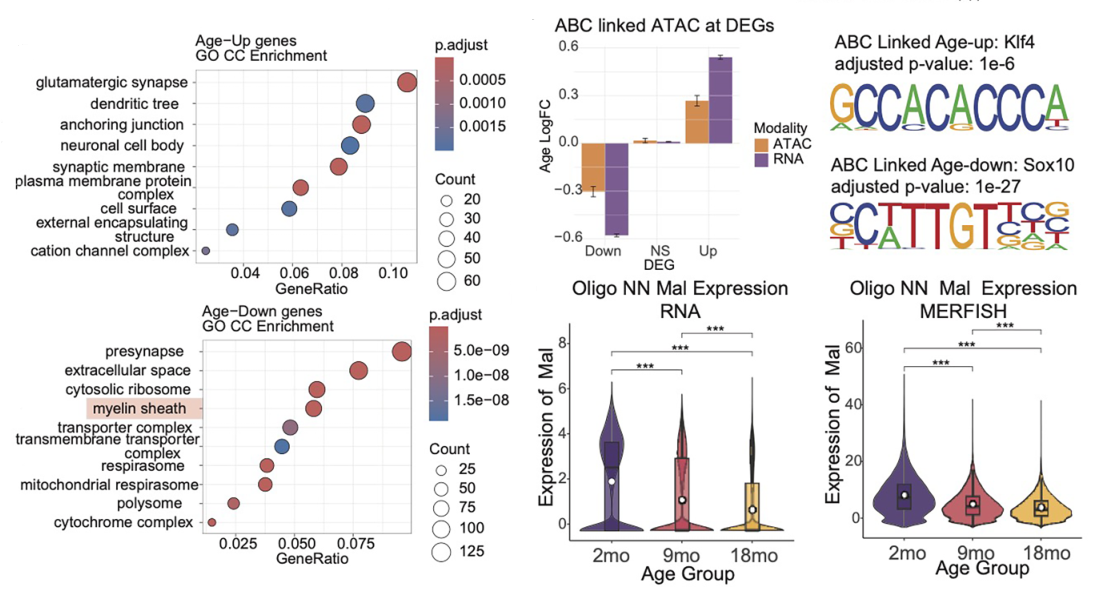
Analysis of the whole-genome DNA methylation data first revealed cell-specific hypomethylation and hypermethylation events. Regions that lost DNA methylation with increasing age in neurons displayed an enrichment for activator protein 1 (AP-1) transcription factor binding motifs, suggesting that AP-1 activation may disrupt cell identity during aging (Patrick et al.). Meanwhile, regions that gained DNA methylation with increasing age in glia displayed enrichment for Sox family transcription factor binding motifs; of note, a loss of Sox activity during aging may impair glial cell function and lead to myelin loss (Stolt et al.). Interestingly, the authors also revealed that DNA methylation patterns at transposable elements distinguished cell types and age groups, and highlighted specific cell types in which the demethylation of transposable elements occurred and promoted increased chromatin accessibility and transposable element transcription, potentially identifying cells susceptible to age-induced neurodegenerative disorders. The authors note that identifying potentially pathogenic transposable element activity in this manner may allow them to distinguish this activity from regulatory transposable element activity and provide a platform for the development of targeted transposable element-silencing interventions that may help to combat the deleterious consequences of aging.
Subsequent analysis of chromatin conformation during aging in the brain at the single-cell level provided what the authors considered the most comprehensive single-cell chromatin conformation dataset on the aging brain to date. These data revealed broad structural remodeling, indicated by the progressive strengthening of the boundaries of topologically associated domains - self-interacting chromosomal regions flanked by CCCTC-binding factor (CTCF) protein binding sites that create insulated neighborhoods essential for gene regulation (Dixon et al.) - and increased chromatin accessibility at CTCF sites. Strengthened topologically associated domain boundaries may function to restrict promoter-enhancer interactions and downregulate the expression of genes that require cross-boundary enhancers, which could negatively affect cell fate and function during aging to a degree that supports the development of neurodegenerative disorders.
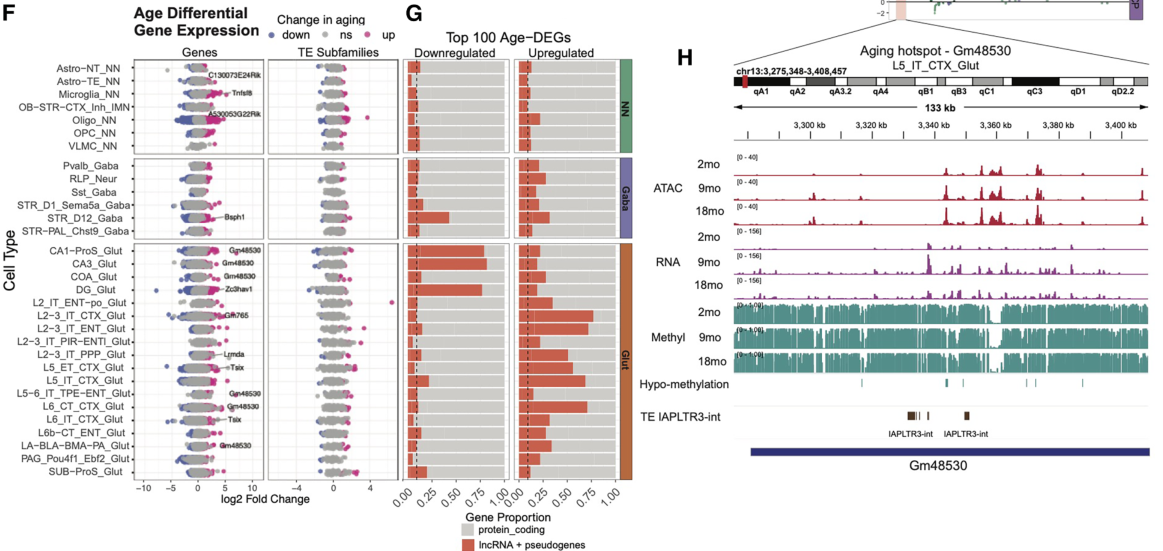
Spatial transcriptomics analysis of nearly 900,000 cells also uncovered regional heterogeneity in aging responses within the same cell types, including altered gene expression in neurons that impact immune responses and involvement of non-neurons as well.
Finally, the authors used the epigenetic and transcriptomic data from this study to train a deep learning model - EpiAgingTransformer (EAT) - that reliably predicts age-related changes in gene expression from multi-modal epigenetic features, providing deep mechanistic insights into gene regulation and highlighting the value of integrative approaches for deciphering brain aging. Interestingly, this model revealed the greater influence of DNA methylation at CpG sites in non-neurons, whereas DNA methylation outside CpG sites and chromatin looping had more prominence in neurons. Finally, the model linked increased chromatin opening to age-associated upregulation of gene expression, but to an alternative regulatory mechanism for downregulated genes residing in constitutively open chromatin regions.
Exploring the Epigenetics of Brain Aging: Could Paired-Tag Lend a Helping Hand?
The findings of this exciting new study help to uncover fundamental aspects of mouse brain epigenetic aging, including: i) the vulnerability of glial cells; ii) cell-type-specific transposable element activation; and iii) increased topologically associated domain boundary strength, which helps to describe the cell-type- and region-specific nature of the normal aging process. Furthermore, the comprehensive single-cell multi-omic atlas of the aging mouse brain reported here will provide a boost to subsequent mechanistic studies and the development of targeted treatment strategies for the all-too-common and often devastating neurodegenerative diseases.
The implementation of Paired-Tag technology from Epigenome Technologies, which generates joint epigenetic and transcriptomic profiles at single-cell resolution and detects histone modifications and RNA transcripts in individual nuclei with efficiency comparable to single-nucleus RNA-seq/ChIP-seq assays, has the potential to provide deeper insight into such research aims. What more could the simultaneous single-cell analysis of histone modification and transcriptomic profiles tell us about the links between aging and the development of neurodegenerative disorders in single cells in crucial regions across the mouse brain?